Drug design
PharmaInformatic has developed novel algorithms in order to generate reliable and accurate models. Complex molecular patterns have been related with specific biological activity of molecules. The use of pharmacophores, 3D superposition or similarity analysis is helpful to detect distinct molecular patterns.
We can model the biological activity of new molecules by using 3D-structures of experimentally defined drug targets (rule based docking, de novo design).
Our rule based docking approach uses deceptively simple rules and methods, which enormously reduce the calculation time in comparison to molecular mechanics or quantum mechanic calculations. Our approach is able to treat all atoms of each target as flexible, e.g. free movement is allowed.
The Protein database (PDB) contains more than 104.500 entries of molecular structures. More than 92.700 have been obtained by X-ray crystallography; about 10.700 were determined by nuclear magnetic resonance (NMR).
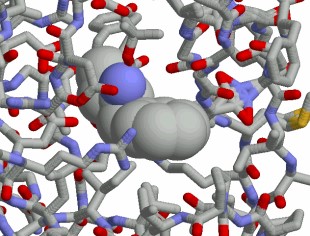
The Protein database provides unique information about the interaction between inhibitors, drugs and biomolecules.
Our methods in structure based drug design are integrated into our platform technology and can be part of our expert systems.

The largest knowledge base
on bioavailability worldwide.

An expert system for finding
suitable
drug candidates.

An expert system for finding molecules with antibiotic activity.

An expert system for finding molecules with antiviral activity.


|
|
|
© Copyright 2004-2021 PharmaInformatic Boomgaarden. All rights reserved. Site map Contact Terms of Use Imprint |
By using our platform technology, we can detect relationships between complex molecular patterns and properties of molecules.
IMPACT-F calculates oral bioavailability in humans much more precisely
than animal trials.
 |
Bioavailability prediction helps to reduces failures
in clinical trials.